马дёҠжіЁеҶҢе…ҘдјҡпјҢз»“дәӨ专家еҗҚжөҒпјҢдә«еҸ—иҙөе®ҫеҫ…йҒҮпјҢи®©дәӢдёҡз”ҹжҙ»еҸҢиөўгҖӮ
жӮЁйңҖиҰҒ зҷ»еҪ• жүҚеҸҜд»ҘдёӢиҪҪжҲ–жҹҘзңӢпјҢжІЎжңүеёҗеҸ·пјҹз«ӢеҚіжіЁеҶҢ


x
еҶңдёҡеҶңжқ‘йғЁеҠһе…¬еҺ…е…ідәҺиҝӣдёҖжӯҘеҒҡеҘҪж–°зүҲе…ҪиҚҜGMPе®һж–Ҫе·ҘдҪңзҡ„йҖҡзҹҘ еҗ„зңҒгҖҒиҮӘжІ»еҢәгҖҒзӣҙиҫ–еёӮеҶңдёҡеҶңжқ‘пјҲеҶңзү§пјүгҖҒз•ңзү§е…ҪеҢ»еҺ…пјҲеұҖгҖҒ委пјүпјҢж–°з–Ҷз”ҹдә§е»әи®ҫе…өеӣўеҶңдёҡеҶңжқ‘еұҖпјҡ
дёәиҝӣдёҖжӯҘеҒҡеҘҪгҖҠе…ҪиҚҜз”ҹдә§иҙЁйҮҸз®ЎзҗҶ规иҢғпјҲ2020е№ҙдҝ®и®ўпјүгҖӢпјҲд»ҘдёӢз®Җз§°ж–°зүҲе…ҪиҚҜGMPпјүиҙҜеҪ»е®һж–Ҫе·ҘдҪңпјҢдҫқжҚ®гҖҠе…ҪиҚҜз®ЎзҗҶжқЎдҫӢгҖӢгҖҒж–°зүҲе…ҪиҚҜGMPе’ҢеҶңдёҡеҶңжқ‘йғЁе…¬е‘Ҡ第292еҸ·жңүе…іиҰҒжұӮпјҢй’ҲеҜ№е®һж–ҪиҝҮзЁӢдёӯеӯҳеңЁзҡ„жҠҖжңҜйҡҫзӮ№зӯүй—®йўҳпјҢжҲ‘йғЁз»„з»Үз ”з©¶еҲ¶е®ҡдәҶгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜз®ЎзҗҶе’Ңе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收жңүе…із»ҶеҢ–иҰҒжұӮгҖӢпјҢзҺ°еҚ°еҸ‘дҪ 们пјҢиҜ·йҒөз…§жү§иЎҢгҖӮ
иҜ·еҗ„зңҒзә§з•ңзү§е…ҪеҢ»дё»з®ЎйғЁй—ЁеҲҮе®һеҠ ејәз»„з»ҮйўҶеҜјпјҢе‘ЁеҜҶйғЁзҪІе®үжҺ’пјҢдёҘж јжү§иЎҢжЈҖжҹҘйӘҢ收ж ҮеҮҶпјҢзЎ®дҝқдёҖдёӘж ҮеҮҶйӘҢеҲ°еә•гҖҒдёҖжҠҠе°әеӯҗйҮҸеҲ°еә•пјӣиҰҒеҲҮе®һеҠ ејәе…ҪиҚҜGMPжЈҖжҹҘе‘ҳз®ЎзҗҶпјҢдҝқйҡңе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收зӯүе·ҘдҪңз»Ҹиҙ№йңҖжұӮпјҢдёҚж–ӯеҠ еҝ«ж–°зүҲе…ҪиҚҜGMPе®һж–ҪжӯҘдјҗгҖӮеҗҢж—¶пјҢжҲ‘йғЁе°ҶжҢҒз»ӯеҠ ејәеҜ№еҗ„ең°е®һж–Ҫж–°зүҲе…ҪиҚҜGMPзҡ„жҢҮеҜјпјҢеҜ№жү§иЎҢж ҮеҮҶдёҚдёҘж јгҖҒиҗҪе®һе·ҘдҪңдёҚеҲ°дҪҚзҡ„еҚ•дҪҚе’ҢдјҒдёҡиҝӣиЎҢйҖҡжҠҘгҖӮ
жң¬йҖҡзҹҘиҮӘеҸ‘еёғд№Ӣж—Ҙиө·ж–ҪиЎҢпјҢгҖҠеҶңдёҡйғЁеҠһе…¬еҺ…е…ідәҺеҚ°еҸ‘гҖҲе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收иҜ„е®ҡж ҮеҮҶиЎҘе……иҰҒжұӮгҖүзҡ„йҖҡзҹҘгҖӢпјҲеҶңеҠһеҢ»гҖ”2013гҖ•26еҸ·пјүеҗҢж—¶еәҹжӯўпјӣжӯӨеүҚзӣёе…іж–Ү件дёӯдёҺжң¬йҖҡзҹҘеҶ…е®№дёҚдёҖиҮҙзҡ„пјҢд»Ҙжң¬йҖҡзҹҘдёәеҮҶгҖӮ
йҷ„件пјҡе…ҪиҚҜз”ҹдә§и®ёеҸҜз®ЎзҗҶе’Ңе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收жңүе…із»ҶеҢ–иҰҒжұӮ
еҶңдёҡеҶңжқ‘йғЁеҠһе…¬еҺ… 2021е№ҙ9жңҲ14ж—Ҙ йҷ„件
е…ҪиҚҜз”ҹдә§и®ёеҸҜз®ЎзҗҶе’Ңе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收жңүе…із»ҶеҢ–иҰҒжұӮ
дёҖгҖҒжҖ»дҪ“иҰҒжұӮ
пјҲдёҖпјүе…ҪиҚҜз”ҹдә§дјҒдёҡеңЁгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжңүж•ҲжңҹеұҠж»ЎеүҚжңӘз”іиҜ·е»¶жңҹжҲ–иҷҪжҸҗеҮәз”іиҜ·дҪҶжңӘз»Ҹжү№еҮҶеҗҢж„ҸгҖҒ并дәҺжңүж•ҲжңҹеұҠж»ЎеҗҺжҸҗдәӨж ёеҸ‘гҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢз”іиҜ·зҡ„пјҢжҢүз…§ж–°е»әдјҒдёҡиҰҒжұӮејҖеұ•е…ҪиҚҜGMPжЈҖжҹҘйӘҢ收пјӣз¬ҰеҗҲ规е®ҡзҡ„пјҢз”ұе®Ўжү№йғЁй—ЁйҮҚж–°зј–еҸ·ж ёеҸ‘гҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢпјҢдјҒдёҡдҫқжі•йҮҚж–°з”іиҜ·ж ёеҸ‘е…ҪиҚҜдә§е“Ғжү№еҮҶж–ҮеҸ·гҖӮ
пјҲдәҢпјүе…ҪиҚҜз”ҹдә§дјҒдёҡиҝҒеқҖйҮҚе»әгҖҒгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжіЁй”ҖжҲ–иў«еҗҠй”ҖгҖҒгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжңүж•ҲжңҹеұҠж»ЎжңӘз”іиҜ·йӘҢ收гҖҒгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжңүж•ҲжңҹеұҠж»ЎеҗҺдёҚеҶҚд»ҺдәӢе…ҪиҚҜз”ҹдә§жҙ»еҠЁпјҢд»ҘеҸҠгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжңүж•ҲжңҹеҶ…зј©е°Ҹз”ҹдә§иҢғеӣҙзҡ„пјҢзңҒзә§з•ңзү§е…ҪеҢ»дё»з®ЎйғЁй—Ёеә”йҖҡиҝҮвҖңеӣҪ家е…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒдҝЎжҒҜз®ЎзҗҶзі»з»ҹвҖқ еЎ«жҠҘдјҒдёҡзӣёе…ідҝЎжҒҜпјҢеңЁзі»з»ҹдёӯжҳҺзЎ®ж–ҮеҸ·жіЁй”ҖиҢғеӣҙ并дёҠдј зӣёе…іиҜҒжҳҺжҖ§ж–Ү件пјҢз”ұжҲ‘йғЁжіЁй”Җзӣёе…іе…ҪиҚҜдә§е“Ғжү№еҮҶж–ҮеҸ·пјҢ并дәҲд»Ҙе…¬е‘ҠгҖӮ
пјҲдёүпјүж–°е»әе…Ҫз”Ёз”ҹзү©еҲ¶е“Ғз”ҹдә§дјҒдёҡгҖҒе…Ҫз”Ёз”ҹзү©еҲ¶е“Ғз”ҹдә§дјҒдёҡйғЁеҲҶз”ҹдә§зәҝеңЁгҖҠе…ҪиҚҜз”ҹдә§и®ёеҸҜиҜҒгҖӢжңүж•ҲжңҹеҶ…д»ҺжңӘз»„з»ҮиҝҮзӣёе…ідә§е“Ғз”ҹдә§д»ҘеҸҠж–°еўһз”ҹдә§иҢғеӣҙзҡ„пјҢж¶үеҸҠзҡ„з”ҹдә§зәҝеқҮйңҖе…ҲйҖҡиҝҮе…ҪиҚҜGMPйқҷжҖҒжЈҖжҹҘйӘҢ收пјҢеҶҚз”іиҜ·еҠЁжҖҒжЈҖжҹҘйӘҢ收пјӣе…¶д»–жғ…еҪўеҸҜзӣҙжҺҘз”іиҜ·еҠЁжҖҒжЈҖжҹҘйӘҢ收гҖӮеұһдәҺжҠ—еҺҹ委жүҳз”ҹдә§зҡ„дҪ“еӨ–иҜҠж–ӯеҲ¶е“Ғз”ҹдә§зәҝпјҲBзұ»пјүпјҢдё”з”ҹдә§иҝҮзЁӢдёҚж¶үеҸҠеҫ®з”ҹзү©зӣёе…іж“ҚдҪңзҡ„пјҢеҸҜзӣҙжҺҘз”іиҜ·е…ҪиҚҜGMPеҠЁжҖҒжЈҖжҹҘйӘҢ收гҖӮ
е…Ҫз”Ёз”ҹзү©еҲ¶е“Ғз”ҹдә§дјҒдёҡеңЁйҖҡиҝҮе…ҪиҚҜGMPйқҷжҖҒжЈҖжҹҘйӘҢ收并иҮӘгҖҠзҺ°еңәжЈҖжҹҘйӘҢ收йҖҡзҹҘд№ҰгҖӢеҮәе…·д№Ӣж—Ҙиө·1е№ҙеҶ…з”іиҜ·еҠЁжҖҒжЈҖжҹҘйӘҢ收зҡ„пјҢеҸӘйңҖжҸҗдҫӣгҖҠе…ҪиҚҜGMPжЈҖжҹҘйӘҢ收申иҜ·иЎЁгҖӢгҖҒиҜ•з”ҹдә§GMPиҝҗиЎҢжғ…еҶөжҠҘе‘Ҡе’Ңдә§е“Ғжү№з”ҹдә§жЈҖйӘҢи®°еҪ•пјӣеҜ№еңЁгҖҠзҺ°еңәжЈҖжҹҘйӘҢ收йҖҡзҹҘд№ҰгҖӢеҮәе…·д№Ӣж—Ҙиө·1е№ҙеҗҺз”іиҜ·еҠЁжҖҒжЈҖжҹҘйӘҢ收зҡ„пјҢжҢүз…§еҶңдёҡйғЁе…¬е‘Ҡ第2262еҸ·з¬¬еӣӣжқЎиҰҒжұӮжҸҗдҫӣе…ЁйЎ№з”іжҠҘиө„ж–ҷгҖӮ
пјҲеӣӣпјүз”ҹдә§иҪҰй—ҙйғЁеҲҶеҠҹиғҪй—ҙгҖҒжЈҖйӘҢз”ЁеҠЁзү©е®һйӘҢе®ӨиҝӣиЎҢж”№йҖ жҲ–дё»иҰҒи®ҫеӨҮеҸ‘з”ҹеҸҳеҢ–пјҢжӯӨзұ»дёҚж¶үеҸҠз”ҹдә§иҢғеӣҙж”№еҸҳдҪҶеҜ№дә§е“ҒиҙЁйҮҸеҸҜиғҪдә§з”ҹйҮҚеӨ§еҪұе“Қзҡ„еҸҳжӣҙпјҢе…ҪиҚҜз”ҹдә§дјҒдёҡеә”еңЁеҸҳжӣҙеҗҺ10дёӘе·ҘдҪңж—ҘеҶ…еҗ‘зңҒзә§з•ңзү§е…ҪеҢ»дё»з®ЎйғЁй—ЁжҸҗдәӨеҸҳжӣҙжғ…еҶөжҠҘе‘ҠпјҢж¶үеҸҠжҙҒеҮҖеҢәж”№йҖ зҡ„иҝҳеә”еҗҢж—¶жҸҗдәӨжҙҒеҮҖжЈҖжөӢжңәжһ„еҮәе…·зҡ„жЈҖжөӢжҠҘе‘ҠгҖӮ
зңҒзә§з•ңзү§е…ҪеҢ»дё»з®ЎйғЁй—Ёеә”еҠ ејәеҜ№еҸҳжӣҙжғ…еҶөзҡ„зӣ‘зқЈжЈҖжҹҘгҖӮе…¶дёӯпјҢж¶үеҸҠж— иҸҢе…ҪиҚҜгҖҒе…Ҫз”Ёз”ҹзү©еҲ¶е“Ғз”ҹдә§иҪҰй—ҙдё»иҰҒеҠҹиғҪй—ҙгҖҒдё»иҰҒз”ҹдә§и®ҫеӨҮжҲ–ж”»жҜ’з”ЁжЈҖйӘҢеҠЁзү©е®һйӘҢе®ӨеҸ‘з”ҹж”№еҸҳзҡ„пјҢзңҒзә§з•ңзү§е…ҪеҢ»дё»з®ЎйғЁй—Ёеә”еңЁж”¶еҲ°еҸҳжӣҙжҠҘе‘ҠеҗҺ20дёӘе·ҘдҪңж—ҘеҶ…еҜ№дјҒдёҡж”№йҖ жғ…еҶөејҖеұ•зӣ‘зқЈжЈҖжҹҘгҖӮ
дәҢгҖҒеҺӮеҢәпјҲеҺӮжҲҝпјүеёғеұҖиҰҒжұӮ
е…ҪиҚҜз”ҹдә§дјҒдёҡеҺӮжҲҝзҡ„йҖүеқҖгҖҒеёғеұҖгҖҒе»әйҖ еә”з¬ҰеҗҲе…ҪиҚҜGMPиҰҒжұӮгҖӮж–°зүҲе…ҪиҚҜGMPе’Ңжң¬ж–Үдёӯе…ідәҺеҺӮжҲҝдёҺе»әзӯ‘зү©зҡ„иЎЁиҝ°дёәеҗҢзӯүеҗ«д№үгҖӮе…·дҪ“иҰҒжұӮеҰӮдёӢпјҡ
пјҲдёҖпјүе…ҪиҚҜз”ҹдә§пјҲе…ҪеҢ»дҪ“еӨ–иҜҠж–ӯеҲ¶е“Ғз”ҹдә§йҷӨеӨ–пјүеә”е…·еӨҮдё“з”Ёзҡ„еҺӮжҲҝпјҢз”ҹдә§еҺӮжҲҝдёҚеҫ—з”ЁдәҺз”ҹдә§йқһе…ҪиҚҜдә§е“ҒгҖӮдҪ“еӨ–иҜҠж–ӯеҲ¶е“Ғжү§иЎҢгҖҠе…ҪеҢ»иҜҠж–ӯеҲ¶е“Ғз”ҹдә§иҙЁйҮҸз®ЎзҗҶ规иҢғгҖӢиҰҒжұӮпјҢеңЁдёҚеҪұе“Қдә§е“ҒиҙЁйҮҸзҡ„еүҚжҸҗдёӢпјҢе…Ғи®ёдҪҝз”ЁеӨҡеұӮеҺӮжҲҝдёӯзҡ„дёҖеұӮжҲ–еӨҡеұӮиҝӣиЎҢз”ҹдә§гҖҒжЈҖйӘҢзӯүжҙ»еҠЁгҖӮ
пјҲдәҢпјүдёҚеҗҢе…ҪиҚҜз”ҹдә§дјҒдёҡдёҚеҫ—еңЁеҗҢдёҖеҺӮжҲҝеҶ…иҝӣиЎҢе…ҪиҚҜз”ҹдә§гҖҒд»“еӮЁгҖҒжЈҖйӘҢзӯүжҙ»еҠЁгҖӮ
пјҲдёүпјүдёҚеҗҢзұ»еһӢзҡ„еҚұйҷ©зү©ж–ҷгҖҒзҗҶеҢ–жҖ§иҙЁдёҚзЁіе®ҡзҡ„зү©ж–ҷеә”йҡ”зҰ»еӮЁеӯҳгҖӮ
дёүгҖҒиҪҰй—ҙеёғеұҖиҰҒжұӮ
еӯҳеңЁеӨҡдә§е“Ғе…ұз”Ёз”ҹдә§иҪҰй—ҙгҖҒи®ҫж–Ҫи®ҫеӨҮзҡ„пјҢдјҒдёҡеә”еҜ№е…ұз”ЁеҸҜиЎҢжҖ§иҝӣиЎҢиҜ„дј°пјҢ并йҡҸзқҖе…ҪиҚҜиЎҢдёҡе’ҢGMPж°ҙе№ізҡ„жҸҗеҚҮпјҢеҜ№е…ұз”Ёж–№ејҸеҸҠж—¶иҝӣиЎҢи°ғж•ҙе’Ңж”№иҝӣгҖӮ
пјҲдёҖпјүеә”жҢүз…§е…ҪиҚҜGMPиҰҒжұӮеҜ№з”ҹдә§иҪҰй—ҙиҝӣиЎҢеҗҲзҗҶеёғеұҖпјҢдёҚеҫ—йҡҸж„Ҹж”№еҸҳжҙҒеҮҖзә§еҲ«пјӣзЎ®йңҖжҸҗй«ҳжҙҒеҮҖзә§еҲ«зҡ„пјҢеә”йҮҮеҸ–жңүж•Ҳзҡ„жҺ§еҲ¶жҺӘж–ҪпјҢйҒҝе…Қдәәзү©жөҒгҖҒз”ҹдә§ж“ҚдҪңеҸҠжё…жҙҒзҒӯиҸҢзӯүеҜ№дә§е“ҒгҖҒзҺҜеўғйҖ жҲҗжұЎжҹ“гҖҒдәӨеҸүжұЎжҹ“д»ҘеҸҠз”ҹзү©е®үе…ЁйЈҺйҷ©гҖӮ
пјҲдәҢпјүжңҖз»ҲзҒӯиҸҢеӨ§е®№йҮҸйқҷи„үжіЁе°„еүӮеә”и®ҫзҪ®дё“з”Ёзҡ„з”ҹдә§иҪҰй—ҙгҖӮ
пјҲдёүпјүжңҖз»ҲзҒӯиҸҢж— иҸҢжіЁе°„еүӮдёҚеҫ—дёҺйқһжңҖз»ҲзҒӯиҸҢж— иҸҢжіЁе°„еүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙпјӣд№іжҲҝжіЁе…ҘеүӮжҲ–еӯҗе®«жіЁе…ҘеүӮдёҚеҫ—дёҺжіЁе°„еүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙпјӣеҸЈжңҚжә¶ж¶ІеүӮдёҚеҫ—дёҺжіЁе…ҘеүӮжҲ–йқһжңҖз»ҲзҒӯиҸҢж— иҸҢжіЁе°„еүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙпјӣеҸЈжңҚеҲ¶еүӮдёҚеҫ—дёҺеӨ–з”ЁеҲ¶еүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙпјӣдёӯиҚҜзүҮеүӮгҖҒдёӯиҚҜйў—зІ’еүӮдёҚеҫ—дёҺзІүеүӮгҖҒйў„ж··еүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙпјӣеҢ–иҚҜзүҮеүӮгҖҒеҢ–иҚҜйў—зІ’еүӮдёҚеҫ—дёҺж•ЈеүӮе…ұз”Ёз”ҹдә§иҪҰй—ҙгҖӮ
пјҲеӣӣпјүеҸЈжңҚжә¶ж¶ІеүӮдёҺжңҖз»ҲзҒӯиҸҢе°Ҹе®№йҮҸжіЁе°„еүӮгҖҒжңҖз»ҲзҒӯиҸҢеӨ§е®№йҮҸйқһйқҷи„үжіЁе°„еүӮеҰӮеӯҳеңЁе…ұз”Ёз”ҹдә§иҪҰй—ҙзҡ„пјҢеҸЈжңҚжә¶ж¶ІеүӮдёҺжіЁе°„еүӮзҡ„з§°йҮҸй—ҙгҖҒй…Қж¶Ій—ҙгҖҒиҚҜж¶Іиҫ“йҖҒз®ЎйҒ“е’ҢзҒҢиЈ…й—ҙеқҮеә”еҗ„иҮӘдё“й—Ёи®ҫзҪ®гҖӮ
пјҲдә”пјүдёӯиҚҜеҸҜжә¶жҖ§зІүзҡ„з”ҹдә§жқЎд»¶йҒөз…§ж•ЈеүӮиҰҒжұӮпјҢеңЁз”ҹдә§иҝҮзЁӢдёӯеә”иҝӣиЎҢеҫ®з”ҹзү©жҺ§еҲ¶гҖӮ
пјҲе…ӯпјүз”ҹдә§жңүеӣҪ家ж ҮеҮҶзҡ„дёӯиҚҜжҸҗеҸ–зү©пјҢе…¶дёӯиҚҜжҸҗеҸ–收иҶҸиҪҰй—ҙеә”е…·еӨҮдёҺжүҖз”ҹдә§жҸҗеҸ–зү©зӣёйҖӮеә”зҡ„еҠҹиғҪй—ҙе’Ңи®ҫж–Ҫи®ҫеӨҮпјҢдёҚеҫ—еңЁдёӯиҚҜеҲ¶еүӮиҪҰй—ҙеҶ…иҝӣиЎҢдёӯиҚҜжҸҗеҸ–зү©зҡ„е№ІзҮҘгҖҒзІүзўҺгҖҒж··еҗҲзӯүж“ҚдҪңгҖӮ
пјҲдёғпјүдёӯиҚҜеҸҜжә¶жҖ§зІүгҖҒз»ҸдёӯиҚҜжҸҗеҸ–еҲ¶жҲҗзҡ„ж•ЈеүӮзӯүдә§е“ҒпјҢе…¶еұһдәҺеҲ¶еүӮз”ҹдә§е·ҘиүәиҢғз•ҙзҡ„зІүзўҺгҖҒж··еҗҲгҖҒеҲҶиЈ…зӯүз”ҹдә§е·ҘеәҸпјҢдёҚеҫ—и®ҫзҪ®еңЁдёӯиҚҜжҸҗеҸ–收иҶҸиҪҰй—ҙеҶ…гҖӮ
пјҲе…«пјүиҙЁйҮҸж ҮеҮҶжңүеҫ®з”ҹзү©йҷҗеәҰжЈҖжҹҘиҰҒжұӮзҡ„дә§е“ҒпјҢе…¶з”ҹдә§зҺҜеўғеә”жҢүз…§Dзә§жҙҒеҮҖеҢәзҡ„иҰҒжұӮи®ҫзҪ®гҖӮ
пјҲд№қпјүз”ЁдәҺе…ҪеҢ»жүӢжңҜеҷЁжў°ж¶ҲжҜ’гҖҒд№іеӨҙжөёжіЎж¶ҲжҜ’д»ҘеҸҠжңүеҫ®з”ҹзү©йҷҗеәҰжЈҖжҹҘиҰҒжұӮзҡ„ж¶ҲжҜ’еүӮпјҢз”ҹдә§зҺҜеўғеә”жҢүз…§Dзә§жҙҒеҮҖеҢәзҡ„иҰҒжұӮи®ҫзҪ®пјӣзҺҜеўғз”Ёж¶ҲжҜ’еүӮеҰӮдёҺйңҖжҙҒеҮҖеҢәз”ҹдә§зҡ„ж¶ҲжҜ’еүӮдә§е“Ғе…ұз”Ёз”ҹдә§иҪҰй—ҙпјҢз”ҹдә§зҺҜеўғеә”жҢүз…§Dзә§жҙҒеҮҖеҢәзҡ„иҰҒжұӮи®ҫзҪ®пјҢ并еә”еҒҡеҘҪзҺҜеўғз”Ёж¶ҲжҜ’еүӮеҜ№Dзә§жҙҒеҮҖеҢәзҡ„жұЎжҹ“жҺ§еҲ¶е’ҢйЈҺйҷ©иҜ„дј°гҖӮ
еӣӣгҖҒи®ҫж–Ҫи®ҫеӨҮиҰҒжұӮ
пјҲдёҖпјүзІүеүӮгҖҒйў„ж··еүӮгҖҒж•ЈеүӮзҡ„з§°йҮҸгҖҒжҠ•ж–ҷеҗҺзҡ„е·ҘеәҸеә”дҪҝз”ЁеҜҶй—ӯејҸи®ҫеӨҮз”ҹдә§пјҢжҲ–йҮҮз”ЁеҜҶй—ӯз®ЎйҒ“гҖҒеҜҶй—ӯејҸ移еҠЁж–ҷд»“зӯүж–№ејҸиҫ“йҖҒзү©ж–ҷпјӣж–ҷж–—зӯүи®ҫеӨҮеӯҳж”ҫеә”дёҺжё…жҙ—еҲҶејҖпјҢжё…жҙ—й—ҙдёҚеҫ—ејҖй—ЁзӣҙжҺҘйҖҡеҗ‘з§°йҮҸжҠ•ж–ҷгҖҒж··еҗҲгҖҒеҲҶиЈ…зӯүеҠҹиғҪй—ҙгҖӮ
пјҲдәҢпјүз”ҹдә§иҝҮзЁӢдёӯдёҚеҫ—еҲ©з”Ёз©әи°ғеӣһйЈҺд»ЈжӣҝйҷӨе°ҳзі»з»ҹпјҢеҜ№дә§е°ҳйҮҸеӨ§зҡ„з”ҹдә§еҢәеҹҹпјҢеә”и®ҫзҪ®дё“з”Ёзҡ„йҷӨе°ҳзі»з»ҹпјҢйҳІжӯўзІүе°ҳжү©ж•ЈпјҢйҒҝе…ҚдәӨеҸүжұЎжҹ“гҖӮ
пјҲдёүпјүж— иҸҢе…ҪиҚҜе’Ңе…Ҫз”Ёз”ҹзү©еҲ¶е“Ғз”ҹдә§иҝҮзЁӢдёӯдә§з”ҹзҡ„еәҹејғзү©еҮәеҸЈдёҚеҫ—дёҺзү©ж–ҷиҝӣеҸЈеҗҲз”ЁдёҖдёӘж°”й”Ғй—ҙжҲ–дј йҖ’зӘ—пјҲжҹңпјүгҖӮ
пјҲеӣӣпјүж— иҸҢе…ҪиҚҜзҡ„зү©ж–ҷеҸ–ж ·дёҚеҫ—йҮҮз”ЁвҖңдёҖиҲ¬еҢә+йҮҮж ·иҪҰвҖқзҡ„ж–№ејҸпјҢеә”еңЁзӣёеә”жҙҒеҮҖзә§еҲ«еҢәеҹҹиҝӣиЎҢеҸ–ж ·гҖӮ
пјҲдә”пјүеә”ж №жҚ®жүҖз”ҹдә§е“Ғз§Қзҡ„жЈҖйӘҢиҰҒжұӮпјҢи®ҫзҪ®з”ЁдәҺж— иҸҢжЈҖжҹҘгҖҒеҫ®з”ҹзү©йҷҗеәҰжЈҖжҹҘгҖҒжҠ—з”ҹзҙ еҫ®з”ҹзү©жЈҖе®ҡд»ҘеҸҠйҳіжҖ§иҸҢж“ҚдҪңзӯүеҫ®з”ҹзү©е®һйӘҢе®ӨгҖӮеҫ®з”ҹзү©е®һйӘҢзҡ„еҗ„йЎ№е·ҘдҪңеә”еңЁдё“еұһзҡ„еҢәеҹҹиҝӣиЎҢпјҢд»ҘйҷҚдҪҺдәӨеҸүжұЎжҹ“гҖӮеҗ„ж“ҚдҪңеҢәеҹҹзҡ„еёғеұҖи®ҫзҪ®гҖҒи®ҫж–Ҫи®ҫеӨҮе’ҢзҺҜеўғзӣ‘жөӢзӯүиҰҒжұӮеә”з¬ҰеҗҲгҖҠдёӯеӣҪе…ҪиҚҜе…ёгҖӢйҷ„еҪ•гҖҠе…ҪиҚҜеҫ®з”ҹзү©е®һйӘҢе®ӨиҙЁйҮҸз®ЎзҗҶжҢҮеҜјеҺҹеҲҷгҖӢгҖҠе…ҪиҚҜжҙҒеҮҖе®һйӘҢе®Өеҫ®з”ҹзү©зӣ‘жөӢе’ҢжҺ§еҲ¶жҢҮеҜјеҺҹеҲҷгҖӢзӯүзӣёе…іи§„е®ҡгҖӮ
дә”гҖҒйӘҢиҜҒдёҺи®°еҪ•иҰҒжұӮ
пјҲдёҖпјүйҮҮз”Ёж— иҸҢз”ҹдә§е·Ҙиүәзҡ„дә§е“ҒпјҲеҰӮйқһжңҖз»ҲзҒӯиҸҢж— иҸҢе…ҪиҚҜгҖҒжңүе…іе…Ҫз”Ёз”ҹзү©еҲ¶е“Ғзӯүпјүеә”иҝӣиЎҢеҹ№е…»еҹәжЁЎжӢҹзҒҢиЈ…иҜ•йӘҢгҖӮ
пјҲдәҢпјүе…ҪиҚҜжү№еҢ…иЈ…и®°еҪ•дёӯеә”жңүжң¬жү№дә§е“ҒиөӢз”өеӯҗиҝҪжәҜз Ғж ҮиҜҶж“ҚдҪңзҡ„иҜҰз»Ҷжғ…еҶөпјҢеҰӮеҢ…иЈ…ж•°йҮҸгҖҒиөӢз Ғи®ҫеӨҮзј–еҸ·гҖҒиөӢз Ғж•°йҮҸгҖҒиҝҪжәҜз ҒдҝЎжҒҜпјҲ24дҪҚж•°еӯ—пјүзӯүпјҢиҝҪжәҜз ҒдҝЎжҒҜд»ҘеҸҠеҜ№дёӨзә§д»ҘдёҠеҢ…иЈ…иҝӣиЎҢиөӢз Ғе…іиҒ”е…ізі»дҝЎжҒҜзӯүи®°еҪ•еҸҜйҮҮз”Ёз”өеӯҗж–№ејҸдҝқеӯҳгҖӮ
йҷ„件пјҡеҶңдёҡеҶңжқ‘йғЁеҠһе…¬еҺ…е…ідәҺиҝӣдёҖжӯҘеҒҡеҘҪж–°зүҲе…ҪиҚҜGMPе®һж–Ҫе·ҘдҪңзҡ„йҖҡзҹҘ.ofd
| 
 еӨҜе®һзІ®йЈҹдә§иғҪ з«Ҝзүўдёӯ
еӨҜе®һзІ®йЈҹдә§иғҪ з«Ҝзүўдёӯ ж–°еҸ‘еұ•йҳ¶ж®өжҺЁеҠЁд№Ўжқ‘жҢҜ
ж–°еҸ‘еұ•йҳ¶ж®өжҺЁеҠЁд№Ўжқ‘жҢҜ еҲҳз…ңиҫүжңҖж–°жј”и®Іе…Ёж–Үпјҡ
еҲҳз…ңиҫүжңҖж–°жј”и®Іе…Ёж–Үпјҡ еҲҳй”ӢпјҡеӯҳйҮҸиө„дә§иҜҒеҲёеҢ–
еҲҳй”ӢпјҡеӯҳйҮҸиө„дә§иҜҒеҲёеҢ–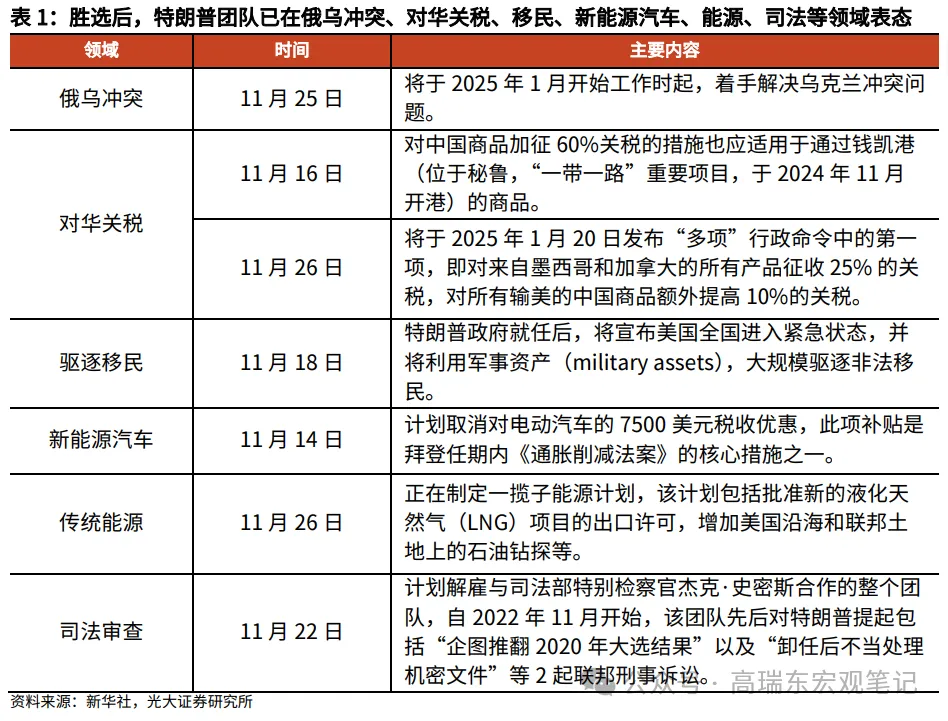 й«ҳз‘һдёңзӯүпјҡд»ҺжӢңзҷ»еҲ°зү№
й«ҳз‘һдёңзӯүпјҡд»ҺжӢңзҷ»еҲ°зү№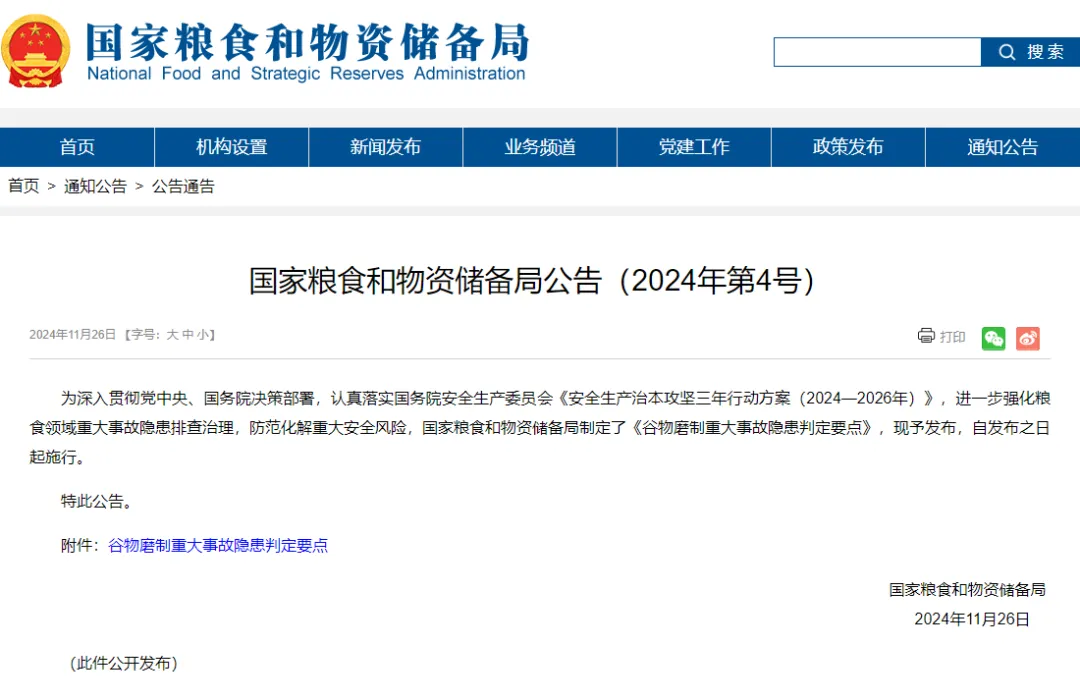 и°·зү©зЈЁеҲ¶йҮҚеӨ§дәӢж•…йҡҗжӮЈ
и°·зү©зЈЁеҲ¶йҮҚеӨ§дәӢж•…йҡҗжӮЈ д»ҳжҢҜеҘҮпјҡ家жҲ·е…ізі»и§Ҷи§’
д»ҳжҢҜеҘҮпјҡ家жҲ·е…ізі»и§Ҷи§’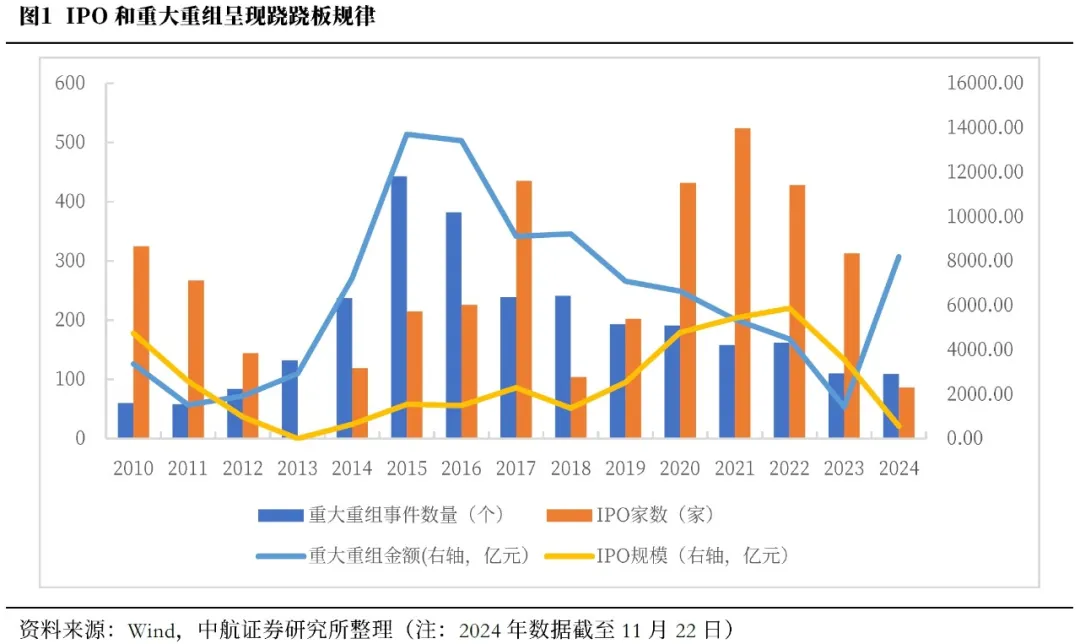 и‘Јеҝ дә‘зӯүпјҡе…іжіЁж–°дёҖиҪ®
и‘Јеҝ дә‘зӯүпјҡе…іжіЁж–°дёҖиҪ® зҪ—еҝ—жҒ’зӯүпјҡеҶ…йҳҒжҲҗе‘ҳжҖқ
зҪ—еҝ—жҒ’зӯүпјҡеҶ…йҳҒжҲҗе‘ҳжҖқ йҖҸи§ҶйғЁеҲҶдё»зІ®иӮІз§ҚвҖңеҗҢ
йҖҸи§ҶйғЁеҲҶдё»зІ®иӮІз§ҚвҖңеҗҢ вҖңдёӯзҫҺеӨ§иұҶиҙёжҳ“йў„жңҹвҖқ
вҖңдёӯзҫҺеӨ§иұҶиҙёжҳ“йў„жңҹвҖқ иұҶзІ•пјҡеҲ©з©әеҮәе°ҪжӢҗзӮ№жңҖ
иұҶзІ•пјҡеҲ©з©әеҮәе°ҪжӢҗзӮ№жңҖ









 еҸ‘иЎЁдәҺ 2021-9-16 10:29:02
еҸ‘иЎЁдәҺ 2021-9-16 10:29:02
 жҸҗеҚҮеҚЎ
жҸҗеҚҮеҚЎ зҪ®йЎ¶еҚЎ
зҪ®йЎ¶еҚЎ ж–°еҸ‘еұ•йҳ¶ж®өжҺЁеҠЁд№Ўжқ‘жҢҜе…ҙйҮҚзӮ№её®жү¶еҺҝдәәжүҚеӣһеј•
ж–°еҸ‘еұ•йҳ¶ж®өжҺЁеҠЁд№Ўжқ‘жҢҜе…ҙйҮҚзӮ№её®жү¶еҺҝдәәжүҚеӣһеј• д»ҳжҢҜеҘҮпјҡ家жҲ·е…ізі»и§Ҷи§’дёӢдј з»ҹеҶңжқ‘зӨҫдјҡжІ»зҗҶзҡ„
д»ҳжҢҜеҘҮпјҡ家жҲ·е…ізі»и§Ҷи§’дёӢдј з»ҹеҶңжқ‘зӨҫдјҡжІ»зҗҶзҡ„ еҰӮдҪ•жһ„е»әејҳжү¬ж•ҷиӮІе®¶зІҫзҘһзҡ„жңәеҲ¶дҪ“зі»пјҹжҠҠжҸЎеҘҪ
еҰӮдҪ•жһ„е»әејҳжү¬ж•ҷиӮІе®¶зІҫзҘһзҡ„жңәеҲ¶дҪ“зі»пјҹжҠҠжҸЎеҘҪ


